Implementation
All the software implementations that we propose (Python, Julia, and C++) offer the same interface from the terminal through the command adabmDCA.
The complete list of training options can be listed through the command
$ adabmDCA train -h
The standard command for starting the training of a DCA model is
$ adabmDCA train -m <model> -d <fasta_file> -o <output_folder> -l <label>
where:
<model>\(\in\) {bmDCA, eaDCA, edDCA} selects the training routine. By default, the fully connected bmDCA algorithm is used. edDCA can follow two different routines: either it decimates a pre-trained bmDCA model, or it first trains a bmDCA model and then decimates it. The corresponding commands are shown below (see section edDCA);<fasta_file>is the FASTA file, with the complete path, containing the MSA;<output_folder>is the path to a (existing or not) folder where to store the output files;<label>is an optional argument. If provided, it will label the output files. This is helpful when running the algorithm multiple times in the same output folder.
Once started, the training will continue until the Pearson correlation coefficient between the two-point statistics of the model and the empirical one obtained from the data reaches a modifiable target value (set by default at --target 0.95).
Output files
By default the training algorithm outputs three kinds of text files:
<label>_params.dat: file containing the parameters of the model saved in this format:Lines starting with
Jrepresent entries of the coupling matrix, followed by the two interacting positions in the sequence and the two amino acids or nucleotides involved.Lines starting with
hrepresent the bias, followed by a number and a letter indicating the position and the amino acid or nucleotide subject to the bias.
Note that inactive, i.e. zero couplings are not included in the file.
<label>_chains.fasta: FASTA file containing the sequences being the last state of the Markov chains used during the learning;<label>_adabmDCA.log: .log file collecting the temporary information of the ongoing procedure.
During the training the output files are overwritten every 50 epochs.
Restoring an interrupted training
It is possible to start the training by initializing the parameters of the model and the chains at a given checkpoint. To do so, two arguments specifying the path of the parameters and the chains are needed:
$ adabmDCA train [...] -p <file_params> -c <file_chains>
Importance weights
It is possible to provide the algorithm with a pre-computed list of importance weights to be assigned to the sequences by giving the path to the text file to the argument -w. If this argument is not provided, the algorithm will automatically compute the weights using Eq.(1) and it will store them into the folder <output_folder> as <label>_weights.dat.
Choosing the alphabet
By default, the algorithm will assume that the input MSA belongs to a protein family, and it will use the preset alphabet defined in Table Alphabets (by default: --alphabet protein). If the input data comes from RNA or DNA sequences, it has to be specified by passing respectively rna or dna to the --alphabet argument. There is also the possibility of passing a user-defined alphabet, provided that all the tokens match with those that are found in the input MSA. This can be useful if one wants to use a different order than the default one for the tokens, or in the eventuality that one wants to handle additional symbols present in the alignment. This is done using --alphabet ABCD- if for example the alphabet contains the symbols A, B, C, D, -.
eaDCA
To train an eaDCA model, we just have to specify –model eaDCA. Two
more hyperparameters can be changed:
--factivate: The fraction of inactive couplings that are selected for the activation at each update of the graph. By default, it is set to 0.001.--gsteps: The number of parameter updates to be performed on the given graph. By default, it is set to 10.
For this routine, the number of sweeps for updating the chains can be typically reduced to 5, since only a fraction of all the possible couplings have to be updated at each iteration.
edDCA
To launch a decimation with default hyperparameters, use the command:
$ adabmDCA train -m edDCA -d <fasta_file> -p <file_params> -c <file_chains>
where <file_params> and <file_chains> are, respectively, the file name the parameters and the chains (including the path) of a previously trained bmDCA model. The edDCA can perform two routines as described above. In the first routine, it uses a pre-trained bmDCA model and its associated chains provided through the parameters <file_params> and <file_chains>. The routine makes sure everything has converged before starting the decimation of couplings. It repeats this process for up to 10000 iterations if needed. If these parameters are not supplied, the second routine initializes the model and chains randomly, trains the bmDCA model to meet convergence criteria, and then starts the decimation process as described. Some important parameters that can be changed are:
--gsteps: The number of parameter updates to be performed at each step of the convergence process on the given graph. By default, it is set to 10.--drate: Fraction of active couplings to be pruned at each graph update. By default, it is set to 0.01.--density: Density of the graph that has to be reached after the decimation. The density is defined as the ratio between the number of active couplings and the number of couplings of the fully connected graph. By default, it is set to 0.02.--target: The Pearson correlation coefficient to be reached to assume the model to have converged. By default, it is set to 0.95. The range of the allowed slope values is fixed to the default interval.
How to choose the hyperparameters?
The default values for the hyperparameters are chosen to be a good compromise between having a relatively short training time and a good quality of the learned model for most of the typical input MSA, where for typical we mean a clean MSA with only a few gaps for each sequence and a not too structured dataset (not too clustered in subfamilies) that could create some ergodicity problems during the training. It may happen, though, that for some datasets some adjustments of the hyperparameters are needed to get a properly trained model. The most important ones are:
Learning rate
By default, the learning rate is set to 0.05, which is a reasonable value in most cases. If the resampling of the model is bad (very long thermalization time or mode collapse), one may try to decrease the learning rate through the argument --lr to some smaller value (e.g. 0.01 or 0.005).
Number of Markov Chains
By default, the number of Markov chains is set to 10000, which works well in most cases. Using fewer chains reduces the memory required to train the model, but it may also lead to a longer algorithm convergence time.
To change the number of chains, we can use the argument --nchains.
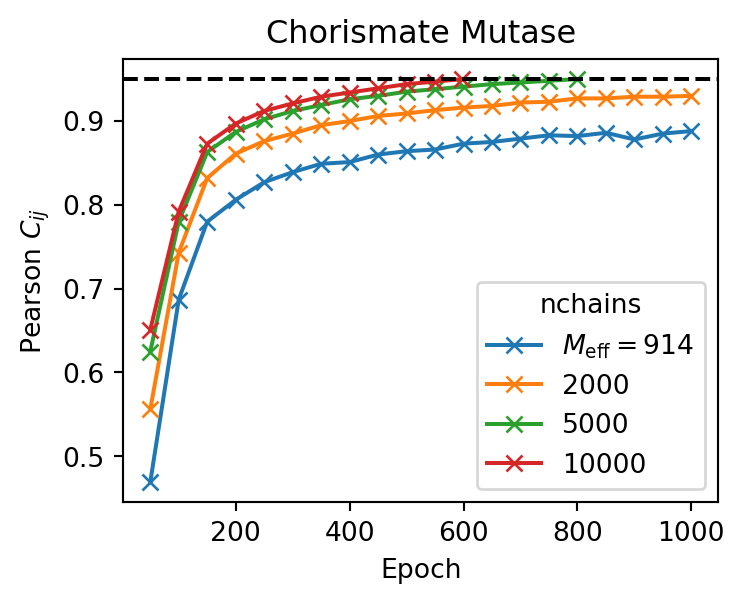
Evolution of the Pearson correlation coefficient for trainings with a different number of Markov chains. The target pearson is set to 0.95 (black dashed line).
Number of Monte Carlo steps
The argument --nsweeps defines the number of Monte Carlo chain updates (sweeps) between one gradient update and the following. A single sweep is obtained once we propose a mutation for all the residues of the sequence. By default, this parameter is set to 10, which is a good choice for easily tractable MSAs. The higher this number is chosen, the better the quality of the training will be, because in this way we allow the Markov chains to decorrelate more to the previous configuration. However, this parameter heavily impacts the training time, so we recommend choosing it in the interval 10 - 50.
Regularization
Another parameter that can be adjusted if the model does not resample correctly is the pseudo count, \(\alpha\), which can be changed using the key --pseudocount. The pseudo count is a regularization term that introduces a flat prior on the frequency profiles, modifying the frequencies as in equations (2).
If \(\alpha = 1\) we impose an equal probability to all the amino acids to be found in the residue at position \(i\), while if \(\alpha = 0\) we just use the empirical frequencies of the data. By default, the pseudo count is set as the inverse of the effective number of sequences, \(1 / M_{\mathrm{eff}}\).